1.1.1 Muscle structure and contraction
The question of muscle regulation is fundamental to our understanding of muscle contraction. The skeletal muscle fiber contains myofibrils which comprise an interdigitated array of thin and thick filaments. The thick filaments contain the protein myosin, which has an elongated structure with two "heads", a neck region which is the binding site for light chains, and a long tail. The tail of myosin is an a helical coiled coil, which interacts in a bundle with other myosin tails to form the bipolar thick filaments. The myosin heads protrude from the surface of the thick filaments, and interact with the thin filaments, each of which comprises a helical array of actin molecules, tropomyosin, and troponin. Muscle contraction occurs when myosin heads interact with actin subunits in a cyclical manner, causing the thin and thick filaments to slide along each other [1,2]. The chemical and mechanical components of this cycle are tightly coupled [3]. The myosin head, an actin activated, Mg2+-ATPase dependent molecular motor, binds to actin and undergoes a power stroke that pulls the thick filament along the thin filament. The myofibrillar array provides an excellent example of a basic principle: The structure of the parts gives rise to the function of the whole. Thus, we can address our fundamental question by examining the structure of the muscle components.
1.2.1 Myosin linked regulation
In many types of muscle, the regulatory switch resides in the thick filament. For example, in vertebrate smooth muscle, myosin is regulated by phosphorylation. Rising Ca2+ levels leads to phosphorylation of the regulatory light chain by myosin light chain kinase. In the on state, the regulatory light chain is phosphorylated [cf.: 4-6]. In molluscan striated muscles, myosin is regulated by Ca2+. The binding of Ca2+ ions to myosin activates contraction [7,8]. Although these biochemical mechanisms are fairly well understood, the structural details of myosin linked regulation are not known at this time. It should be noted that phosphorylation can also have a modulatory effect on tension in skeletal muscle [9], where regulation is under thin filament control.
1.2.2 Thin filament regulation
Regulation of contraction in vertebrate skeletal and cardiac muscles depends on the thin filament proteins, troponin and tropomyosin. Tropomyosin (Tm) is a two stranded helical coiled coil that bonds head to tail to form filaments which wind in the grooves of the actin helix. Troponin (Tn), associated with Tm in a 1:1 ratio, consists of three subunits: calmodulin-like TnC binds Ca2+; TnI inhibits the actomyosin ATPase; and TnT anchors troponin to Tm. Each Tm/Tn complex spans and regulates 7 actin monomers. In the relaxed state, TnI binds strongly to actin, so that Tm is held in a blocking position which covers the myosin binding sites on the actin filament. When the Ca2+ concentration rises to activating levels, the subunit interactions within the troponin complex are strengthened, so that the interaction of TnI with actin is weakened, and Tm is released from its blocking position. Tm then moves deeper into the actin groove where it is weakly bound to actin by specific recognition sites on the molecule. Myosin then interacts effectively with actin, and contraction commences [10,11].
This
mechanism of thin filament regulation, known as the steric blocking mechanism,
is based on the x-ray diffraction measurements, which can detect structural
transitions within the thin filament of skeletal muscle. More recently,
Ca2+ dependent movement of Tm in the thin filament has been
observed in three dimensional reconstruction electron micrographs [12].
It has been shown that Tm blocks access to the myosin binding sites on
the actin filament, and moreover, that Tm moves away from these blocking
sites in the presence of activating levels of Ca2+ [13]. Cooperative
action might be expected in this Ca2+ dependent transition,
because of the head to tail linkage of the tropomyosin molecules along
the thin filament. There is some experiment support for such cooperative
effects [14].
TnC, the calcium sensitive switch
TnC is representative of the calmodulin (CAM) superfamily of intracellular calcium receptors. The light chains of myosin are also members of this protein family. High resolution structures of skeletal 2Ca2+TnC [15,16] revealed a dumbbell-shaped molecule composed of two globular lobes, connected by a long helical linker, the so-called central helix. This dumbbell shape is now known to be typical of the protein family [17-20]. Each lobe comprises two Ca2+ binding domains known as EF hands (Figure 1.1). An EF hand comprises a Ca2+ binding loop which connects two helices [21]. Although the members of this protein family share similar structures, they have different physiological roles. Thus, any additional structural information which can be obtained about them may have broad biological implications in the study of cellular signaling and motility.
Herzberg
et
al. proposed that the binding of Ca2+ to TnC leads to the
opening of two EF hands, which exposes the hydrophobic interior of the
regulatory N terminal lobe (N lobe) and provides a binding site for TnI
[22]. In the "closed" and "open" states of the N lobe, the individual
EF hands can be characterized by their interhelical angles. In the closed
state, the interhelical angle (A-B or C-D angle) is about 131° or more,
while in the open state, the interhelical angle is about 115° or less.
The proposed N lobe conformational change was observed by NMR [23] and
in the high resolution structures of 4Ca 2+TnC [17], and Ca2+
saturated N lobe of TnC [18]. The structures illustrated in atomic detail
the alterations of specific interactions that result in the conformational
shift and they also explained some of the functional differences between
the structural C terminal lobe (C lobe) and the calcium sensing N lobe.
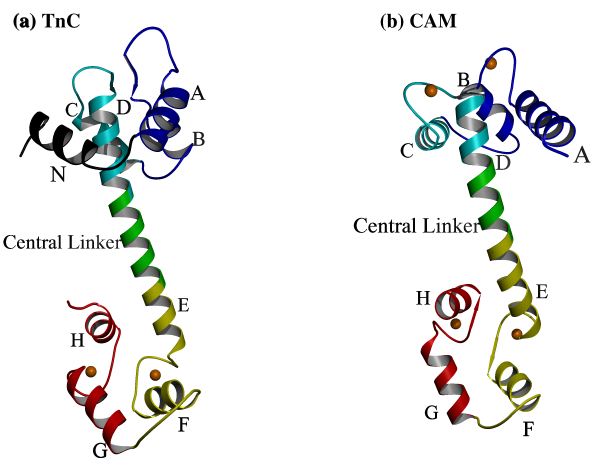
Figure
1.1.
X-ray crystallographic structures of (a) TnC [15] and (b) CAM [20] share a similar structural fold; two globular domains (lobes) held separate by an extended, helical central linker. Each lobe comprises two Ca2+ binding EF hands joined by a linker. Each EF hand consists of two a helices joined by a Ca2+ binding loop (the eight helices are named A-H, from the N terminus to the C terminus). In TnC there is an additional helix (helix N) N terminal to helix A which is not present in CAM. The so-called "central helix" comprises helix D, the central linker, and helix E. In skeletal TnC and in CAM, all four EF hands are able to bind Ca2+, but in cardiac TnC (not shown), domain II is unable to bind Ca2+. High resolution structures have shown that the bound Ca2+ is seven-coordinated in a roughly pentagonal bipyramidal arrangement [15-20] (see Figure 2.4).
CAM
and TnC are strikingly similar molecules (Figures 1.1 and 1.2), but there
are important structural and functional differences. As mentioned previously,
the C lobe of TnC plays a structural role in that it anchors the molecule
to TnI. The N lobe of TnC is the calcium switch, which controls regulation.
By contrast, both lobes of CAM are "regulatory" calcium switches. Under
physiological conditions, TnC remains anchored to the thin filament, but
CAM is a solution protein which can bind to and regulate many other proteins
(Figure 1.2). These structural and functional differences arise from certain
differences in the amino acid sequence of the two proteins (see chapter
2).
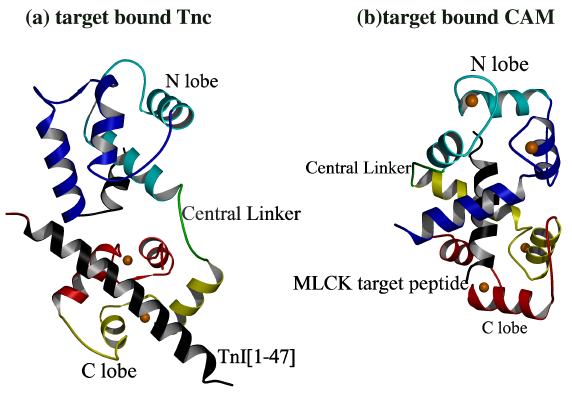
Figure 1.2.
(a) Vassylyev et al. Solved the structure
of 2Ca2+TnC bound to a peptide comprising residues Gly1-His47
of TnI [43]. The complex revealed the structural interaction of TnI with
the hydrophobic core of the TnC C lobe. In the TnC/TnI complex, TnI(Gly1-His47)
provides the "scaffolding" that anchors TnC to the thin filament. Although
the structure has a relatively compact conformation, it is not as compact
as that of (b) calmodulin bound to its target region from myosin light
chain kinase [48]. The N lobe of calmodulin is open and the hydrophobic
core is turned inward towards the target peptide. By contrast, the N lobe
of TnC is closed and the hydrophobic core is turned away from the target
peptide. This difference of interaction illustrates key functional differences
between CAM and TnC. For example, in figure (a), TnC is in the 2Ca2+
off state, but it remains anchored to TnI. By contrast in (b), CAM must
be in the 4Ca2+ on state in order to interact with MLCK peptide.
Based on these structures, it appears that the central helix melts in central
linker region when there are peptide interactions which bring the lobes
close together.
Cardiac TnC, a drug target
In cardiac
muscle, TnC is a potential drug target in the treatment of congestive heart
failure. In contrast to that of skeletal muscle troponin C, the first
loop of cardiac TnC (cTnC) is unable to bind calcium due to the mutation
of Ca2+ ligands (D29L, D31A) and a V28 insertion [24-26].
The structure of cardiac TnC (cTnC) bound to the "calcium sensitizing drug"
bepridil has recently been solved [114]. It has been suggested that
bepridil and the inhibitory region of TnI could bind to the N lobe simultaneously
[27]. In such a case, rational drug design would require an understanding
of any interactions of bepridil with TnI as well as TnC.
TnC-TnI interactions
The Ca2+ dependent interactions between TnC and TnI would be an important component of the steric blocking mechanism, and these interactions have been studied using TnI peptides. The TnC binding domain of TnI is known to consist of two discontinuous regions: The N terminal region consists of residues Gly1-His47. The C terminal region comprises the inhibitory region (TnI residues Gly104-Arg115) [28] and consists of residues Asn96-Arg115 plus some additional C terminal residues [29]. Moreover, TnI(Asn96-Ser117) can bind to actin and thereby inhibit actomyosin ATPase [30], so that the C terminal region appears to be part of the regulatory switch. Various studies indicate that TnC binds to TnI in an anti-parallel manner: the C lobe of TnC interacts with the N terminal residues of TnI, and the N lobe of TnC interacts with the C terminal region [31,32,33,34]. The minimum peptide sequence from TnI which is able to inhibit actomyosin ATPase was determined to be residues Gly104-Arg115 [28], here called the inhibitory region. Peptide TnI(Asn96-Met116), interacts with the N lobe of TnC in a Ca2+ dependent manner [35]. Interactions between peptide TnI(Asn96-Met116) and the C lobe were also observed [36,37], but these interactions were not dependent on Ca2+ [35,38,39]. Moreover, photocrosslinking experiments have shown that the C lobe may not interact with TnI(Asn96-Met116) when whole proteins are used [40]. Affinity measurements and NMR studies have demonstrated that residues C terminal to the inhibitory region are required for Ca2+ dependent interaction with the regulatory N lobe of TnC [41,37]. Thus, it appears that while peptides comprising TnI residues Gly104-Arg115 are sufficient to bind actin, inhibit actomyosin ATPase, and interact with the C lobe of TnC as well, these residues by themselves are not sufficient to represent the Ca2+ dependent interaction of TnI with the N lobe of TnC.
The N terminal region of skeletal TnI (residues Gly1-His47) was predicted to form a basic, amphiphilic helix in its interaction with TnC [42]. This prediction was confirmed by the structure of skeletal 2Ca2+TnC bound to peptide TnI(Gly1-His47) [43]. In that structure the central helix of TnC is not extended, and the central linker of TnC, also known as the DE linker, is unwound (Figure 1.2). TnI(Gly1-His47) is not involved directly in regulation; instead, it anchors the C lobe of TnC to the thin filament in a manner which is not dependent on Ca2+ concentration. The complex is relatively compact, and there are extensive contacts between the N and C lobes. The peptide binds primarily to the C lobe, but also has interactions with the N lobe, which add stability to the compact conformation of the overall complex. The peptide does not interact with the core of the N lobe which turns away from the center of the molecule (Figure 1.2). A model, based on the structural similarity between the two lobes, was proposed for the interaction of the inhibitory region of TnI with TnC [43].
Until recently, no structural information was available about the Ca2+ dependent interaction of TnC with the inhibitory region of TnI. A peptide comprising the inhibitory region has been shown to interact in a non-helical conformation with TnC [44]. NMR experiments showed the somewhat extended conformation of peptide TnI(Lys105-Arg115) in the presence of TnC [45]. NMR experiments also demonstrated that the binding of peptide TnI(Asn96-Thr148) to the TnC N lobe results in a somewhat less open lobe [46]. Moreover, chemical shifts indicated that peptide TnI(Asn96-Thr148) binds extensively to the hydrophobic surface of EF hands 1 and 2, but the specific interactions of TnI with TnC remained unknown. Then, an NMR study of the complex of cardiac troponin I (cTnI) peptide, cTnI(Arg147-Lys163), bound to the N lobe of cTnC showed that the peptide interacts as a helix with EF hand 1 primarily [47]. cTnI(Arg147-Lys163) corresponds to skeletal TnI(Arg115-Lys131), and the sequence is highly homologous. Thus, it is likely that the NMR structure also represents the structure of the N lobe of skeletal TnC bound to its respective peptide.
1.3 Thesis prelude: 4Ca2+TnC with and without peptide
In this thesis I describe our work directed toward answering the following questions; What is the structural mechanism of the Ca2+ switch in TnC, and how does the switch affect the interaction of TnC with TnI? Many studies have addressed this question as detailed in the previous sections. Until the current study, no one had seen the open conformation of 4Ca2+TnC in atomic detail, so it was difficult to understand the precise working of the Ca2+ switch. Although the overall conformational change had been described in the proposal of Herzberg et al. [22], and confirmed by NMR [23], many questions regarding the structural mechanism and the specific role of certain residues remained unanswered. For example; Which residues constitute the "hinge region"? How does the conformational change affect the stability of the central linker? How do sequence differences between CaM and TnC give rise to the different functional properties of these proteins? In order to answer such questions, we have used protein crystallography to examine the structure of skeletal TnC in the 4Ca2+, on state. The results of this study are described in Chapter 2.
Recently, the structure of the bepridil-4Ca2++cTnC complex has been solved in our lab. I provided guidance to Yu Li during the crystallization of the complex, the collection of the data, and the solving and refinement the structure. I also assisted in the writing and editing of the paper. In the structure, both lobes of cTnC are open, and the central linker is fully unwound. This observation provided support for Ca2+ dependent flexibility in the N terminal turn of the central linker of TnC, a notion first proposed in Chapter 2, which now appears to be correct.
In
Chapter 3 we report the first crystal structure of four calcium-bound recombinant
skeletal TnC complexed with a synthetic peptide, TnI(Asn96-Lys123), which
includes the inhibitory region of rabbit skeletal TnI. Before this
structure was solved, the specific Ca2+ dependent interactions
of the inhibitory region with TnC were unknown. In the complex, TnC assumes
an extended conformation with a fully helical central linker. The
peptide is for the most part non-helical in structure, and stabilizes the
N terminal turn of helix in the central linker. In Chapter 3 we also
describe the implications of the structure for the rational design of calcium
sensitizing drugs, and propose a mechanism for TnC/TnI interaction which
may also represent key features of the "on" state of thin filament based
regulation.