with a troponin I peptide [Asn96-Lys123]
In
order to explore the Ca2+-dependent interactions between TnC
and TnI, we have solved the structure of skeletal 4Ca2+TnC co-crystallized
with a synthetic peptide, TnI[Asn96-Lys123]. Here we describe the
implications of the structure for the rational design of calcium sensitizing
drugs, and propose a mechanism which attempts to describe the TnC/TnI interaction
in the "on" state of thin filament based regulation.
3.1 Overall structure description
The
overall conformation of TnC in the complex is similar to that of uncomplexed
4Ca2+TnC [17] with an r.m.s.d. of 2.014 Å for backbone
superimposition. As in the uncomplexed structure, the long central
helix is fully extended such that the N and C lobes are held separate.
Ca2+ is bound to each of the four EF hand domains. The
hydrophobic core of the C lobe is fully exposed to the solvent. By
contrast, that of the N lobe is shielded by residues Leu111-Asp119 of the
peptide. The rest of the peptide residues are disordered. Helices
B, C and D of TnC form a slot which constitutes the core hydrophobic interaction
surface for the peptide strand (Figure 3.1). The peptide stabilizes
the conformation of the N-lobe by forming a bridge between Gln 85 and Asp86
of helix D and the BC linker (Figure 3.2). Moreover, the sidechains
of Arg113 and Arg115 form critical salt bridges with Glu54 of helix C and
Asp86 of helix D, respectively. These interactions orient the N-terminal
region of the peptide as it protrudes from the hydrophobic core of the
N lobe. Peptide residues Arg112-Arg115 constitute a turn which is
stabilized by the interaction of O 112 with the BC linker main chain at
N 49, and by the internal van der Waals interactions between Leu111 and
Val114 of the peptide. This turn of peptide is similar in its overall
conformation to that obtained in an NMR study [81]. In the current structure,
it appears that the interactions of the N terminus of the peptide with
the N-lobe are possibly disrupted by the strong packing interactions of
helix C. There are extensive van der Waals interactions between the peptide
and hydrophobic core of TnC, especially for residues Met116-Ser117 of the
peptide. These core interactions stabilize the open conformation
of the N lobe in general. Together, Ile58 of helix C and the helical groove
of helix D (residues Met78, Arg81, and Gln82) comprise a small target
binding pocket in EF hand 2 which is occupied by the sidechain of Met116
of the peptide. The hydrophobic surfaces of Ser117 interact with
sidechain of Met 79 on helix D and with Met43 and Leu39 of helix B .
O of Ser117 is unpaired, but it does have a close van der Waals interaction
with Leu39. The peptide strand is stabilized by the interaction between
N 116 and Oe 82 of helix D.
The C terminal region of the peptide also follows the helical groove of
helix D and protrudes from the TnC hydrophobic core. The C terminal
region of the peptide is stabilized by an interaction between Od
86 of helix D and N 119. The extensive interaction
of the peptide with Asp86 is consistent with affinity measurements of the
interaction of TnI segments with TnC mutant (E85A/D86A). The authors also
found that the calcium dependent interaction of TnC with TnI depends on
TnI residues C terminal to Met116 [90]. Moreover, TnC mutants which had
deletions in this part of the central linker (Glu85-Ala87), were unable
to fully activate actomyosin ATPase. Thus, it appears that these residues
of the central linker and their correct interaction with TnI are critical
for the transition to the troponin on state.
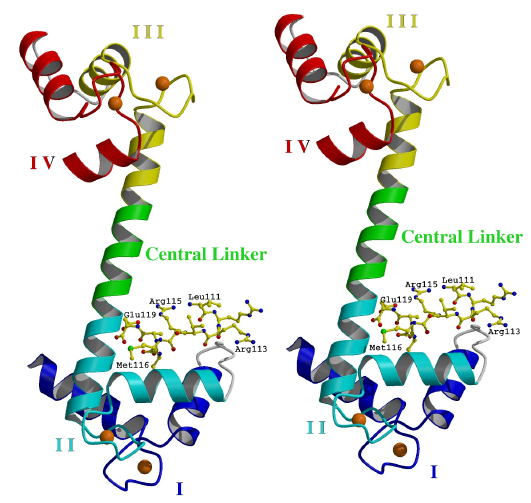
Figure 3.1
Overall structure of TnC/peptide complex
In the complex, TnC (shown in ribbon representation) has calcium ions (spheres) bound to its four EF hand domains (EF hands). Each EF hand consists of two a-helices joined by a calcium-binding loop. The peptide (shown as ball and sticks) binds specifically to the open N lobe. The EF hands are numbered I-IV from the N terminus. Helices A and B are found in EF hands I. Helices C and D are found in EF hands II. Helices E and F are found in EF hands III. Helices G and H are found in EF hands IV. The N terminal helix N is disordered. (Stereo diagram generated using the program Raster3D [115].)
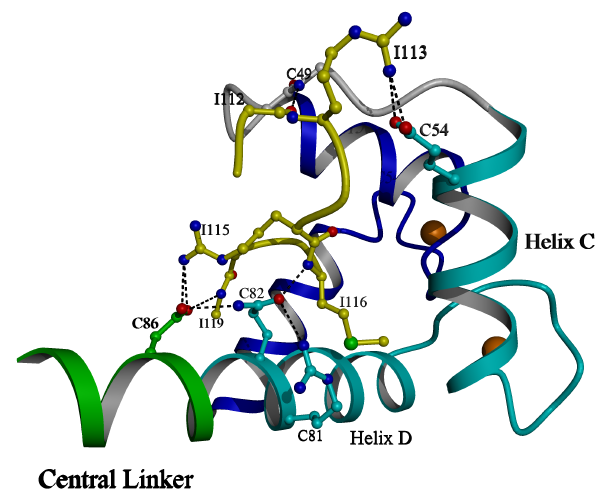
Figure 3.2
Peptide bound to the N lobe
The N-lobe and peptide of the TnC/peptide
complex is shown. The peptide is shown as balls and sticks. The N terminal
region of the peptide, Leu111-Arg115, interacts with helix C, the BC linker
and helix D. The central region of the peptide, Met116-Ser117, interacts
with the hydrophobic core of the N lobe. The C terminal region of
the peptide, Ala118-Asp119, interacts with the helix D groove. Calcium
ions are shown as spheres bound to the loops of EF hands 1 and 2. (Image
generated using the program POVscript [116].)
3.2 Comparison to other
CAM superfamily members
The
N-lobe of uncomplexed 4Ca2+TnC [17] is more open than that of
the current peptide bound structure. It appears that helices B and
D clamp upon the peptide, because it is narrow, non-helical and extended
in its conformation (Table 3.1).
Table 3.1
Torsional character
of peptide backbone angles
LEU
111 turn
ARG 112 turn
ARG 113 turn
VAL 114 turn
ARG 115 turn
MET 116 strand
SER 117 strand
ALA 118 link
ASP 119
This TnI-dependent conformational difference was also observed in a recently published NMR structure which showed the conformation of the N lobe, but not the interactions of the peptide [46]. In contrast to the crystal structure of skeletal TnC complexed with peptide TnI(Gly1-His47) [43], the hydrophobic residues of the peptide in the current structure do not insert deeply into the core at the center of the lobe, and a small cavity is found there, enclosed by Met116 and Ser117 of the peptide, and by Met78, Ile58, Leu39 and Met43 of TnC. Interestingly, there is a similar cavity in the N lobe hydrophobic core of CAM bound to the myosin light chain kinase peptide [48], which is another case were a target peptide does not insert deeply into the lobe.
For
TnC, the hydrophobic faces of the lobes do not turn inward in the current
structure, nor in that of TnC bound to peptide TnI(Gly1-His47) [43], but
the structures of CAM bound to various targets revealed a more compact
conformation with both lobes turned inward toward the target peptide [103,48,52,104].
It is likely that this difference of interaction is related to the distinctive
functions of the two proteins. CAM is found in solution in the cellular
milieu and has a variety of targets, whereas TnC remains anchored to its
sole target, TnI. TnC has a single regulatory lobe, but both lobes
of CAM are regulatory. On the other hand, CAM might also be found
in an extended conformation under particular conditions where one lobe
remains anchored to the target at all times. For example, CAM remains bound
to phosphorylase B kinase g chain even at low
Ca2+ concentrations [94]. CAM is
expected to adopt an extended conformation when bound to the phosphorylase
B kinase g chain, which
shares some sequence similarity with TnI in the CAM binding regions [94].
The mechanism of this Ca2+independent interaction
is not known at this time, but it is likely that the CAM C lobe, which
does not close fully at low calcium [54,64], remains
anchored to phosphorylase B kinase g
chain at all times. For TnC, the Ca2+ independent interaction
with TnI(Gly1-His47) depends
on the high affinity cation binding domains of the
C-lobe, which open in response to either Mg2+ or Ca2+
binding. Thus, the C-lobes of both TnC and CAM
(in this particular case) exhibit Ca2+ independent interaction
with their respective targets, although the mechanism of this Ca2+
independent interaction is likely to be different for CAM and TnC. Moreover,
because of the sequence similarity in the target regions, the current structure
may be representative of the interaction between CAM and phosphorylase
B kinase g chain.
3.3 Stabilization of the central linker by peptide
interactions
3.3.1 Packing considerations
One
of the most striking features of the current structure is the stabilization
of the helical central linker by the peptide (Figure 3.3). Spin labeling
studies have shown that the binding of cTnI(86-211) to cTnC decreases the
flexibility of the central linker and maintains cTnC in an extended conformation
[89]. This is consistent with x-ray and neutron scattering results which
showed that, in the presence of Ca2+, TnC assumes an extended
conformation in its interaction with TnI, with a maximum linear dimension
of about 72 Å [91]. By contrast, the 2Ca2+TnC/TnI(Gly1-His47)
complex [43] has a maximum linear dimension of less than 60 Å.
In the current structure, helix D has packing interactions with loop 4
of the adjacent protomer. This protomeric loop 4 also provides the
central linker with its only packing interaction: The sidechain of
Lys88 of the central linker interacts with the sidechains of Asp147 and
Asp149 of the adjacent protomer. The central linker is otherwise
fully exposed to the large solvent channels. In fact, the central
linker forms a helical pillar which holds apart the layers of lobes in
the xz planes of the triclinic crystal so that the relative disposition
of the independent layers is determined by the disposition of the central
linker (Figure 3.4). Thus, it appears that the stability the crystal
depends on the stability of the central linker in its extended helical
conformation.
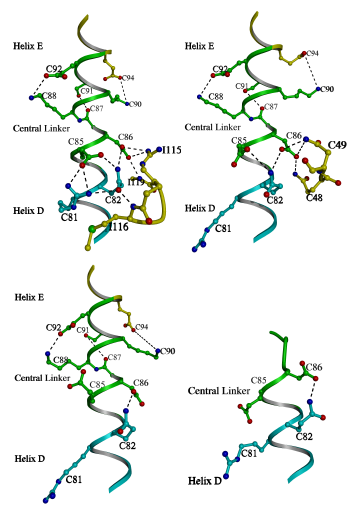
Figure 3.3
A comparison of the central linker stabilization
in various crystal structures of TnC
In the current structure (top left) the N
terminal portion of central linker is constrained by its interaction with
the peptide (Rabbit skeletal numbering is used thoughout for consistency).
These constraints lead to a strengthening of the intrahelical interactions
of the central linker (Table 3.2). The central linker of 2Ca2+TnC
[15] (top right) is constrained in a similar way by the BC linker and the
intrahelical interactions are also strengthened. Correspondingly, the central
helix has a similar disposition overall in the current structure and in
that of 2Ca2+TnC. By contrast, the central linker of 4Ca2+TnC
(bottom left) does not receive as much stabilization [17], and the intrahelical
interactions of the linker are weakened (Table 3.2). The various crystal
forms of 4Ca2+TnC have dissimilar bends in the central helix
[112]. In the complex of 2Ca2+TnC and peptide TnI(Gly1-His47)
[43] (bottom right) the central linker is unwound due to the interactions
of the peptide. Although the N lobe is in the 2Ca2+ state, the
linker stabilizing interactions of Asp86 and Glu85 with the BC linker are
broken. Thus, it appears that the central linker may play a role in regulation,
because its stability is coupled to the state of the N lobe.
Table 3.2
Intrahelical central linker interactions
TnC/peptide 2Ca2+TnC 4Ca2+TnC1 4Ca2+TnC2
Oe94-Nz90(Å) 4.10 5.12 6.86 8.40
Oe92-Nz88(Å) 3.33 3.46 3.04 7.30
OHg91-O87(Å) 3.25* - 4.77 4.19
OHg91-O88(Å) - 2.85 4.04 5.08
3.3.2 Ca2+ dependent central linker flexibility
A
comparison of 4Ca2+TnC and 2Ca2+TnC led to the prediction
of Ca2+ dependent flexibility in central linker in Chapter 2.
This prediction was based not only on the crystallographic structures,
but also on the results from an NMR study [57] and on the structures of
other members of the CAM superfamily [48,58,60,67,68]. These additional
studies indicated the precise regions of unwinding in the central linkers
of various members of the CAM superfamily. Based on this comparison, and
on Ca2+ dependent interaction of the BC linker with the central
linker, also described in Chapter 2, it appeared that the central linker
would be more flexible in the 4Ca2+ on state than in the 2Ca2+
off state. Moreover, a Ca2+ dependent unwinding of the
central linker at residues Lys84-Asp86 was predicted. Although this prediction
received some support from the other structures, it seemed at the time
to border on dangerous speculation. Perhaps we were unaware that
NMR studies had shown a Ca2+ dependence of central linker flexibility
[89]. This prediction has received additional experimental support
in the three years since the initial publication of Chapter 2 in Structure
[17]. For example, a comparison of more recent crystallographic structures
of 4Ca2+TnC reveals general variability in the disposition of
the central helix (although the linker is still fully helical) [112], in
contrast to that of 2Ca2+TnC.
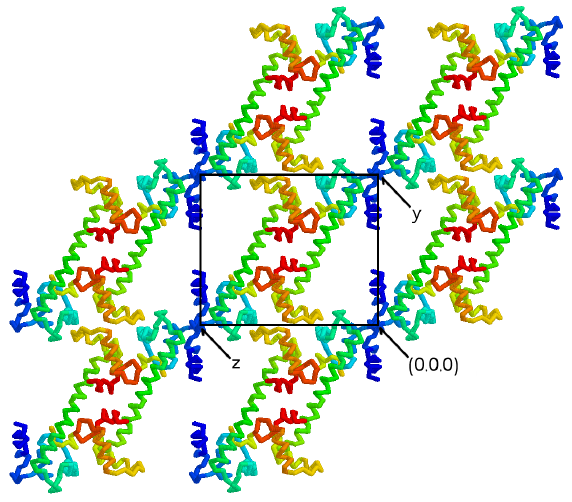
Figure 3.4
The central helix supports layers of lobes
in the crystals
The C2 dimer forms the asymmetric unit of
the P1 cell. Two-fold symmetry is still present in the individual
yz planes of the P1 crystals shown here. The central helix forms
a helical pillar which holds apart the layers of lobes in the xz planes
of the P1 crystal form. In fact, the relative disposition of the
xz planes depends on the stability of the central linker, because no other
interactions bridge the layers. Thus, it appears that the stability
of the crystals depends on the integrity of the helical central linker
in the TnC/peptide complex. (Figure generated using the program RASMOL
[117])
In the structure
of 2Ca2+TnC bound to peptide TnI(Gly1-His47) (Figure 1.2) [43],
the central linker is unwound and the overall conformation of the complex
is relatively compact, because the peptide interacts with both the N and
C lobes. Similarly, the structure of the bepridil-4Ca2++cTnC
complex, also shows that the central linker unwinds when there are interactions
that hold both lobes together [114]. By contrast, the N-lobe of the
bepridil-4Ca2++cTnC complex is open, but the N-lobe of 2Ca2+TnC
bound to peptide TnI(Gly1-His47) is closed, so that the central linker
receives some stabilization from van der Waals interactions with the BC
linker and helix B. In accordance with the prediction, N terminal
central linker residues Lys84-Glu85 are helical, and Asp86 forms single
helical interaction. In this case, Asp86 has a relatively high B factor,
because it is destabilized by the adjacent linker region which is unwound.
In the bepridil-4Ca2++cTnC complex, these residues are unwound
in accordance with the prediction. This Ca2++ dependent flexibility
points to a possible structural role for the central linker of TnC in thin
filament regulation.
3.3.3 Intrahelical stabilization of the central linker
Ever since
the crystal structure of 2Ca2+TnC was first shown to contain
a fully helical central linker [96, 97], the possibility of internal stabilization
within the central helix has been discussed [98]. If the peptide
stabilizes the helical conformation of the central linker, then it could
be expected that these internal stabilizing interactions would also be
strengthened due to the helical dipole [17]. In the N lobe of 2Ca2+TnC
[15], these central linker stabilizing interactions of the peptide are
mimicked by the BC linker of TnC (Figure 3.3). Thus, it would be
expected that internal stabilization of the helical central linker would
also be observed in the 2Ca2+TnC crystal structure. By
contrast, the intrahelical stabilizing interactions of the central linker
would be weaker in the crystal structure of 4Ca2+TnC, where
the central helix receives less stabilization [17]. Moreover, central
linker stabilization by the BC linker would be weaker in cases where the
central linker is non-helical, such as in solution, or in the crystal structure
of 2Ca2+TnC bound to peptide TnI(Gly1-His47) [43]. This
destabilization may facilitate the binding of the inhibitory region of
TnI, because these interactions must be broken to open the lobe [17]. Table
3.2 shows that central linker intrahelical interactions are better overall
for the current complex and 2Ca2+TnC than for 4Ca2+TnC.
Figure 3.3 shows that the various crystal structures are in good agreement
with this notion of central linker stabilization. It has been suggested
that the phi/psi rotation of Gly89 would be a first step in the unwinding
of the central linker [17]. It now appears that these intrahelical
interactions provide some stabilization against unwinding at Gly89 under
conditions where the central linker receives additional stabilization at
Gln85 and Asp86 (Figure 3.3). Moreover, the various crystal structures
of TnC together show that the stabilization of the central linker depends
on the state of the N lobe (Figure 3.5, Table 3.2).
3.3.4 The CAM analogy
It
has been suggested that increased flexibility of the central linker could
improve the interaction of TnI with 4Ca2+TnC because, by analogy,
the central linker is unwound in every known peptide bound structure of
CAM [17,103,48,52]. Accordingly, TnC might interact with TnI in a
less extended conformation than that observed in the 2 or 4Ca2+TnC
crystallographic structures. As more structures have appeared, the
facts have become clearer. In the peptide bound complexes of CAM and in
structure of 2Ca2+TnC bound to peptide TnI(Gly1-His47), the
central linker is unwound because the peptide stabilizes close contacts
between the lobes [43,103,48,52]. A similar situation exists for the bepridil-4Ca2++cTnC
complex [114]. By contrast, in the complex of TnC with peptide TnI(Asn96-Lys123),
the peptide can stabilize a less flexible, helical central linker that
holds the lobes apart.
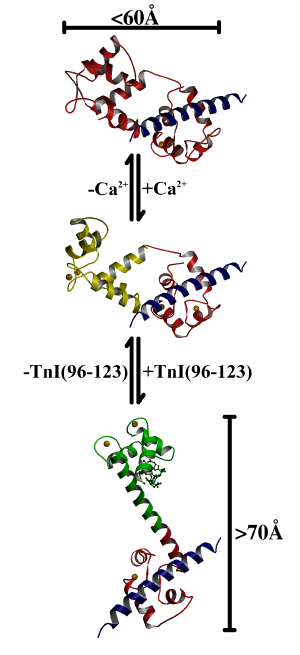
Figure 3.5
A mechanism of interaction between TnC
and TnI
In the proposed mechanism, step 1 is represented by the complex of 2Ca2+TnC and peptide TnI(Gly1-His47) [43]. TnC is not extended and the central linker is unwound. In step 2, calcium ions bind to the N lobe, and it opens and becomes receptive to interaction with the inhibitory region of TnI. In step 3, the inhibitory region of TnI binds and stabilizes the helical conformation of the central linker and the extended conformation of the complex.
3.3.5 The central linker in solution
NMR studies
find (in contrast to the crystal structures) that the central linker melts
and is non-extended when TnC is isolated in solution [95,60]. (There
is a similar inconsistency with respect to the interhelical angles of the
crystal and NMR structures; see Chapter 2). This troublesome fact
can be explained by noting that these are solution structures, where there
is greater freedom of movement. In contrast to TnC, CAM is a solution
protein, and the central linker of CAM is also known to unwind in solution
[57,58]. It is notable that the central linker of CAM has also been observed
as helical in crystal structures [49]. Thus it appears that the extended
conformation of CAM or TnC is not stable in solution. In this sense, crystallographic
structures may better reflect the structure of TnC in the native environment,
because like the thin filament, crystals are protein arrays which can apparently
stabilize the extended conformation of protein molecules.
3.3.6 Implications of TnI interactions with the central linker
In previous structures of uncomplexed TnC helix D is stabilized by helix N, but in the current structure, helix D and the central linker are stabilized by the peptide. There are extensive van der Waals interactions between helix D and peptide which were described earlier. Also, an interlocking chain of interactions between the peptide and TnC that constrain the side chains of helix D and the central linker at Gln85 and Asp86 (Figure 3.3), so that TnI residues Leu111-Asp119 provide exceptional stabilization to the central linker of TnC.
A possible implication of the current structure is that the central linker is extended and helical in the on-state of thin filament regulation. Such an argument finds some support in experiments which suggest a role for the central linker in regulation [101,108,109,110] and in energy transfer experiments on the TnC/TnI complex in the presence of Ca2+ [40]. Regulation may require the added specificity implied by the many TnC/peptide interactions in the current structure. For example, the TnI point mutation Arg113Gly has been implicated in hypertrophic cardiomyopathy (HCM) [105,106,107]. This mutation leads to an increase in Ca2+ sensitivity [106,113]. As noted above this arginine makes a critical salt bridge to Glu54 of helix C (Figure 3.2). In the mutant, the steric constraints on this region of TnI are likely to be weaker, so that TnI would bind to TnC at lower Ca2+ levels. It should be noted that this mutation is
also likely
to affect the interaction of TnI with actin and to impair regulation [113].
The current structure only provides a probable explanation of the Ca2+
sensitivity effect. In another example, the mutation of Asp86 of the central
linker to alanine results in reduced affinity for the peptide [41] and
in defective actomyosin ATPase activation in avian TnC [111]. As described
above, Asp86 makes many contacts with the peptide and is critical for the
stabilization of the central linker. Thus, it is likely that the highly
specific central linker stabilizing interactions in the current structure
are representative of those in the troponin on state.
3.4 TnC/TnI interaction mechanism
As mentioned previously, the central linker has different dispositions in the current structure, and in that of 2Ca2+TnC bound to peptide TnI(Gly1-His47) [43]. Based on this observation, a mechanism for the calcium-dependent interaction of TnC with TnI can be proposed which employs these structures. The first step is represented by the TnC/TnI(Gly1-His47) crystal structure [43] (Figure 3.5). In step 2, when Ca2+ binds, the N lobe simply opens. The overall conformation of TnC is still relatively compact, but the N lobe is receptive to the calcium dependent interaction with TnI (Figure 3.5). Although this structure has not been directly observed, the N lobe interactions with peptide TnI(Gly1-His47) have been detected in the presence of Ca2+ [92]. In step 3, the extension of the central helix probably accompanies the binding of TnI to the N lobe for reasons explained above (Figure 3.5). It should be noted that a fully helical central linker is not required for full extension of the central helix. In other words, it is likely that the N and C lobes could still be held apart by their interaction with TnI, even if the central linker remains partially unwound. Nevertheless, it now seems likely that the added stability of the central linker in the presence of TnI(Leu111-Asp119) would be exploited in the TnC/TnI complex. In a final step, when Ca2+ is released from EF hands 1 and 2, TnI is simply expelled from the closing N lobe. If the extended conformation of TnC is no longer stabilized by TnI, the central linker would unwind and TnC would resume its step 1 conformation. The overall structural disposition of TnC in this final step is unclear at present, but if it were known, a regulatory mechanism would be implied by it.
This
mechanism implies a large conformational change in TnI which would accompany
the rotation and displacement of the N lobe in step 3. There is an
expected displacement of over 25Å for the loop between helices A
and N with respect to loop 4 of the C lobe and the N-terminus of TnI (Figure
3.5). In intact thin filaments, the inhibitory region of TnI would
likely be held away from its actin binding site in the on state, represented
in step 3. These observations are roughly consistent with the results
of energy transfer experiments which showed that Cys133 of TnI moves 15
Å away from Cys374 of actin and 12.6 Å closer to TnC on Ca2+
binding [99, 102]. It is tempting to suggest that this interaction
mechanism represents the conformational switch of thin filament regulation,
however, this suggestion is not supported by our results. The current
structure shows only the Ca2+ dependent interaction of the N
lobe of TnC with a short region of the peptide representing TnI(Asn96-Lys123).
Although this structure cannot represent fully the TnC/TnI interaction
nor its role in regulation, nonetheless, we have shown that these
interactions may represent important aspects of the regulatory role of
TnC/TnI interaction in vertebrate striated muscle.
3.5 Implications for the design of drugs targeting TnC
It
has recently been shown that the deep cavity of the N lobe hydrophobic
core is the interaction site in cTnC for the phenyl ring of bepridil, the
calcium sensitizing drug [114]. It was also proposed that cTnI and
bepridil could bind to the cardiac N-lobe simultaneously [89]. It
is likely that the complex of cTnC and cTnI resembles the current structure.
An overlay of the bepridil-bound cTnC N lobe with the current structure
(Figure 3.6) does produce a clash between (Met116-Ser117) of the peptide
and the isobutoxy chain of bepridil. This clash is probably not significant
because the isobutoxy chain is quite flexible. Thus, the suggestion
appears to be plausible that N lobe of cTnC can accommodate both cTnI and
bepridil. In such a case, the "wedge-like" insertion of bepridil
into the N lobe would result in a more fully open N lobe, and the backbone
interaction of Arg144 of cTnI (sTnI Arg112 equivalent) with the BC linker
of cTnC (sTnC mainchain N 49 equivalent) would likely be broken.
To compensate, hydrogen bonds could be formed between the positively charged
pyrrolidine groups of bepridil and several carbonyl oxygens of N-terminus
of the peptide (Figure 3.6). Thus, an important implication of this comparison
is that bepridil interacts not only with TnC but with TnI as well.
This is consistent with the observed effect of bepridil, the modulation
of contraction in cardiac muscle. Furthermore, modifications of bepridil,
such as the substitution or addition of more basic amphipathic groups for
the pyrrolidine group and the truncation of the isobutoxy chain, may produce
a more powerful calcium sensitizer. Additionally, cTnI and the drug
bind to largely different surfaces in the N lobe, so a drug binding surface
can be mapped, which takes into account the TnC/TnI interaction.
For example, the molecules "Bep1" and "Bep3" can be used to construct an
adductive "model drug" (Figure 3.6), which may be a more specific calcium
sensitizer, enhancing contraction. Alternatively, bulkier compounds
which obstruct the TnI binding surface may attenuate the contractile response.
Such an agent might be useful in the treatment of excessive calcium sensitivity
in diseases such as hypertrophic cardiomyopathy [105,106,107].
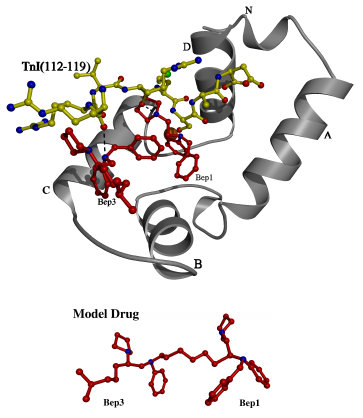
Figure 3.6
An overlay showing possible interactions
of cTnI with cTnC and bepridil
An overlay of the A and D helices was used to model the peptide of the
current structure into the N lobe of cardiac TnC (medium grey) with bepridil
(dark) bound. Some possible interactions are shown with dotted lines
between the carbonyl groups of the peptide and the nitrogens of bepridil.
The sidechain of Bep1 clashes with the mainchain of the peptide, but this
clash is not likely to be significant because the sidechain is quite flexible,
and should be displaced by the peptide (bepridil sidechain not shown).
It appears that the drug and the peptide bind to different surfaces of
the cTnC N lobe, and that the bound bepridils define a drug binding surface
on the N lobe. A polycarbon linkage between Bep1 and Bep3 was used
to construct the "model drug".
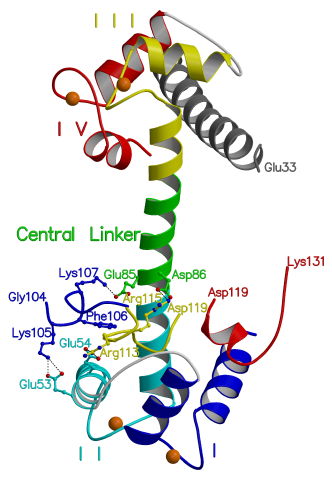
Figure 3.7
Four structures overlayed: Interactions
of TnI with TnC
This model was produced by fitting the shared
coordinates of the following structures to those of the current structure;
TnI(Gly104-Arg115) [45] (blue) bound to EF hand II, TnI(Glu3-Glu33) [43]
(grey) bound to EF hands III and IV, cTnI(Arg147-Lys163) [47] (red) shown
with skeletal numbering and bound to EF hand I. The four EF hands of TnC
are numbered with roman numerals and TnI(Leu111-Asp119) (yellow) is shown
as in the current structure, bound to EF hands I and II. The shared regions
of the overlayed structures are not shown. In the model residues TnI(Lys105-Lys107)
make plausible interactions with EF hand II of TnC in accordance with crosslinking
studies [31,32,118]. These interactions do not appear in the current structure
due to packing constraints around helix C. Residues TnI(Arg115-Ala118,
skeletal TnI numbering) of the cardiac TnC/TnI(Arg147-Lys163) NMR structure
[47] are not shown due to a clash with the current structure in the core
binding region. This clash is probably due to the fact that these residues
are not stabilized by cTnC(Asp86, rabbit skeletal numbering) in the NMR
structure because the central linker is truncated and unwound [47].
3.6 Materials and Methods
We have determined to 2.0 Å resolution the crystal structure of expressed rabbit 4Ca2+TnC grown in the presence of a synthetic peptide comprising TnI residues Asn96-Lys123. Two crystal forms have been obtained: monoclinic (C2) with unit cell dimensions a=109.40 Å, b=26.54 Å, c=59.67 Å, b= 93.44° and triclinic (P1) with unit cell dimensions a=26.28 Å, b=55.29 Å, c=63.26 Å, a=89.53° b=91.46° g=103.72° Crystals of both forms were grown by seeding under identical conditions at 4° C from a mixture of 2 µl of reservoir buffer (50 mM cacodylate pH 6.5, 10 mM CaCl2, 29% ethanol, 29% MPD). The partial specific volume is similar for both crystal forms (~2.8 Å3/Da), with one and two molecules per asymmetric unit for the C2 and P1 crystal forms respectively. Data were collected from the monoclinic crystal form at -180° C to 3.5 Å resolution using a MAR image plate detector mounted on a Rigaku X-ray generator equipped with Supper focusing mirrors. Data for the triclinic crystal form were collected to a resolution of 2.0 Å at -160° C using synchrotron radiation at CHESS (beamline A1,l =0.935 Å). Data reduction was carried out with the programs DENZO [77] and SCALEPACK [78] Table 3.3 summarizes the statistics of diffraction.
The
program AMoRe [85] was used to orient and position the N and C lobes of
4Ca2+TnC independently in the monoclinic asymmetric unit. The
final correlation coefficient after fitting was 44.4 (R factor 47.4) for
all the data between 8 and 3.5 Å. After 400 cycles of refinement
with the program XPLOR [81] the R factor fell to 0.349. A comparison of
the unit cell parameters for the monoclinic and triclinic crystal forms
suggested that the C2 TnC dimer could be used as a search model in AMoRe
for the P1 cell. The final correlation coefficient after fitting was 48.8
(R factor 46.2). After one round of simulated annealing with NCS restraints,
the R factor fell to 0.330. F0-Fc
maps indicated peptide binding specifically to the N lobe but not to the
C lobe. The program O [83] was used to model the peptide into this density
and to improve the fitting of the TnC molecules to the electron density
map (Figure 3.8). The solvent structure was modeled automatically with
the program ARP [82]. The NCS restraints were removed in a final
round of maximum likelihood refinement with the program CNS [93].
The P1 protomers in the final structure are nearly identical with an rms
displacement of 0.140 Å and 0.076 Å for the protein and peptide
respectively. Table 3.4 gives the refinement statistics for both
crystal forms.
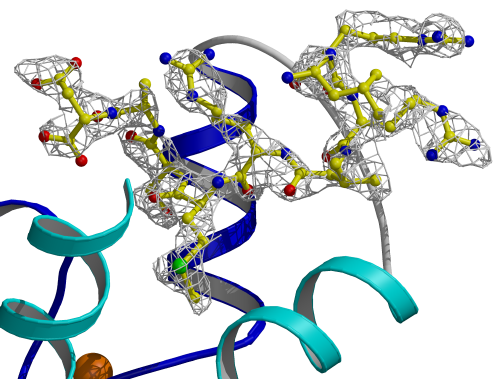
Figure 3.8
Electron density map for TnI[Leu111-Asp119]
The final peptide map was calculated using
the program DM [79] with non-crystallographic symmetry averaging.
Table 3.3
Data collection statistics
Crystal form monoclinic triclinic
Resolution
range (Å) 15.0 - 3.50
50.0 - 2.00
No. of reflections
6573
193252
No. unique reflections
2231
21185
Completeness
97.08
90.9
Rmerge (%)
8.8
7.1
Table 3.4
Refinement statistics
monoclinic triclinic
Number
of reflections
2225
21183
Sigma cutoff
none
none
R factor (%)
29.6
21.8
Free R Factor (%)
33.4
25.8
Number of water molecules
0
131
Rms bond lengths (Å)
0.035
0.008
Rms bond angles (°)
2.744
1.436
Rms bond dihedrals (°)
25.284
20.919
Rms bond impropers (°)
4.085
0.832
Global G factor
-1.0
0.4
Before we commenced this work, the only TnC structure that was known was that of 2Ca2+TnC [15,16]. With its closed N lobe, this structure represents the off state. Based on this structure, and on a comparison to CAM, Herzberg, et al. proposed that the N lobe would open in the presence of Ca2+[22]. It was impossible to describe the structural role of TnC in thin filament regulation based on this one structure, so we first set out to crystallize TnC in the 4Ca2+, on state. The structure of 4Ca2+TnC validated the proposal of Herzberg et al. [22], and revealed at atomic resolution the hinge regions and critical linkages that change in the transition from closed to open. We were also able to show how sequence differences give rise to many of the diverse functional properties of proteins in the CAM superfamily.
Another insight derived from the structure of 4Ca2+TnC was the prediction of Ca2+ dependent flexibility in the central linker, which arises from TnC specific interactions between the BC linker and the central linker in the 2Ca2+ state. In light of the work described in Chapter 3, this now appears as a critical insight. Recently, the structure of the bepridil-4Ca2++cTnC complex has been solved [114]. In the structure, both lobes of cTnC are open, and the central linker is unwound. We have shown in Chapter 3 that a comparison of the bepridil bound structure to that of 2Ca2+TnC bound to peptide TnI(Gly1-His47) [43] provides the first unambiguous crystallographic support for the predicted Ca2++ dependent flexibility in the central linker.
In Chapter
3, the analysis of the interaction between 4Ca2+TnC and TnI(Asn96-Lys123)
suggests the following interpretation of this Ca2+ dependent
flexibility in central linker; Although the central linker of 4Ca2+TnC
is more flexible than that of 2Ca2+TnC, this increase in flexibility
can be reversed by the Ca2+ dependent interaction of TnI with
4Ca2+TnC. Our structure shows that TnI provides stabilization
to a less flexible and helical central linker similar to that observed
in 2Ca2+TnC (17). In particular, TnI residues Arg115-Asp119
provide stabilization to the N terminal turn of helix in the central linker.
This N terminal turn of helix (residues Lys84-Asp86 of sTnC) comprises
residues known to be crucial for regulatory activation [101,41]. In Chapter
3, we have shown that the cruciality of these central linker residues is
attributable to their specific interaction with TnI(Arg115-Asp119).
Moreover, because these interactions appear to be part of the regulatory
switch, the stabilization in TnC of an extended helical central linker
by TnI is probably an important feature of the troponin on state. We have
also shown how a comparison of the current structure to that of bepridil-4Ca2++cTnC
may guide the development of new drugs that are specific to the troponin
complex.
References
1. Huxley, A. F., Niedergerke, R. M.: 173:971-973. (1954)
2. Huxley, H. E., Hanson, J.: Nature. 173:973-976. (1954)
3. Lymn, R. W., Taylor, E. W.: Biochemistry. 10:4617-4624. (1971)
4. Ruegg, J. C.: Calcium in muscle activation: a comparative approach, Springer-Verlag, Berlin, Heidelberg, New York. (1986)
5. Sellers, J. R., Adelstein, R. S., Regulation of contractile activity, The Enzymes XVIII, Academic Press, Orlando and London, pp. 381-418. (1987)
6. Ashley, C. C., Mulligan, I. P., Lea, T. J.: Ca2+ and activation mechanisms in skeletal muscle. Quarterly Review of Biophysics. 24:1-73. (1991)
7. Ohtsuki, I., Maruyama, K. Ebashi, S.: Regulatory and cytoskeletal proteins of vertebrate and skeletal muscle. Advances in Protein Chemistry. 38:1-67. (1986)
8. Szent-Gyorgyi, A. G., Szentkiralyi, E. M., Kendrick-Jones, J. J. Mol. Biol. 74:179-203. (1980)
9. Metzger JM, Greaser ML, Moss RL.: Variations in cross-bridge attachment rate and tension with phosphorylation of myosin in mammalian skeletal muscle fibers. Implications for twitch potentiation in intact muscle. J Gen Physiol. May;93(5):855-83. (1989)
10. Phillips GN Jr, Fillers JP, Cohen C. Tropomyosin crystal structure and muscle regulation. J Mol Biol. 192:111-31. (1986)
11. Squire, J. M., Morris, E. P.: A new look at thin filament regulation in vertebrate skeletal muscle. FASEB. J. 12:761-771. (1998)
12. Lehman W, Craig R, Vibert P.: Ca(2+)-induced tropomyosin movement in Limulus thin filaments revealed by three-dimensional reconstruction. Nature. 68:65-7. (1994)
13. Xu C, Craig R, Tobacman L, Horowitz R, Lehman W.: Tropomyosin positions in regulated thin filaments revealed by cryoelectron microscopy. Biophys J. 77:985-92. (1999)
14. Moss RL, Allen JD, Greaser ML.: Effects of partial extraction of troponin complex upon the tension-pCa relation in rabbit skeletal muscle. Further evidence that tension development involves cooperative effects within the thin filament. J Gen Physiol. 87:761-74. (1986)
15. Herzberg, O. & James, M. N. G.: Refined crystal structure of troponin C from turkey skeletal muscle at 2.0 Å resolution. J. Mol. Biol. 203, 761-779 (1988).
16. Satyshur, K. A., Rao, S. T., Pyzalska, D., Drendel, W., Greaser, M. & Sundaralingam, M. : Refined structure of chicken skeletal muscle troponin C in the two-calcium state at 2-Å resolution. J. Biol. Chem. 263, 1628-1647 (1988).
17. Houdusse, A., Love, M. L., Dominguez, R., Grabarek, Z. & Cohen C. : Structures of four Ca2+-bound troponin C at 2.0 Å resolution: further insights into the Ca2+-switch in the calmodulin superfamily. Structure 5, 1695-1711 (1997).
18. Strynadka, N. C., Cherney, M., Sielecki, A., Li, M. X., Smillie, L. B., James M. N. G.,: Structural details of a calcium-induced molecular switch: X-ray crystallographic analysis of the calcium-saturated N-terminal domain of troponin C at 1.75 Å resolution. J. Mol. Biol. 273, 238-255 (1997).
19. Babu, S. Y., Sack, J. S., Greenhough, T. J., Bugg, C. E., Means, A. R., Cook, W. J: Three-dimensional structure of calmodulin. Nature. 315, 37 (1985).
20. Taylor, D. A., Sack, J. S., Maune, J. F., Beckingham, K., Quiocho, F. A.: Related Articles, Protein, Structure Structure of a recombinant calmodulin from Drosophila melanogaster refined at 2.2-A resolution. J Biol Chem. 266:21375-80. (1991)
21. Kretsinger, R. H., Nockolds, C. E.: Carp muscle calcium-binding protein. II. Structure determination and general description. J. Biol Chem 248:3313-26 (1973)
22. Herzberg, O., Moult, J., James, M. N. G.: A Model for the Ca2+ induced Conformational Transition of Troponin C. J Biol Chem. 261, 2638. (1986)
23. Gagné, S. M., Tsuda, S., Li, M. X., Smillie, L. B. & Sykes, B. D. Structures of the troponin C regulatory domains in the apo and calcium-saturated states. Nat. Struct. Biol. 2, 784-789 (1995).
24. Zot, A.S., Potter, J.D., Strauss, W.L.: Isolation and sequence of a cDNA clone for rabbit fast skeletal muscle troponin C. Homology with calmodulin and parvalbumin. 1987. J. Biol. Chem. 262, 15418.
25. Wilkinson, J.M.: The amino acid sequence of troponin C from chicken skeletal muscle. 1976. FEBS Lett. 70, 254.
26. Wilkinson, J.M.: Troponin C from rabbit slow skeletal and cardiac muscle is the product of a single gene. 1980. Eur. J. Biochem. 103, 179.
27. Kleerekoper, Q., Liu, W., Choi, D. & Putkey, J. A. Identification of binding sites for bepridil and trifluoperazine on cardiac troponin C. J. Biol. Chem. 273, 8153-8160 (1998).
28. Talbot, J. A., Hodges, R. S.: Comparative studies on the inhibitory region of selected species of troponin-I. The use of synthetic peptide analogs to probe structure-function relationships. J. Biol. Chem. 256, 2798-2802 (1981)
29. Syska H., Wilkinson J. M., Grand R. J., Perry S. V.: (1976) The relationship between biological activity and primary structure of troponin I from white skeletal muscle of the rabbit. Biochem J 153:375-87
30. Mornet, D., Bonet-Kerrache, A., Strasburg, G. M., Patchell, V. B., Perry, S. V., Huber. P. A., Marston, S. B., Slatter, D. A., Evans, J. S., Levine, B. A.: The binding of distinct segments of actin to multiple sites in the C-terminus of caldesmon: comparative aspects of actin interaction with troponin-I and caldesmon. Biochemistry. 34:1893-901. (1995)
31. Leszyk, J., Grabarek, Z., Gergely, J., Collins, J. H.: Characterization of zero-length cross-links between rabbit skeletal muscle troponin C and troponin I: evidence for direct interaction between the inhibitory region of troponin I and the NH2-terminal, regulatory domain of troponin C. Biochemistry. 29:299-304. (1990)
32. Kobayashi, T., Tao, T., Grabarek, Z., Gergely, J., Collins, J. H.: Cross-linking of residue 57 in the regulatory domain of a mutant rabbit skeletal muscle troponin C to the inhibitory region of troponin I. J Biol Chem. 266:13746-51. (1991)
33. Krudy, G. A., Kleerekoper, Q., Guo, X., Howarth, J. W., Solaro, R. J., Rosevear, P. R.: NMR studies delineating spatial relationships within the cardiac troponin I-troponin C complex. J Biol Chem. 269:23731-5. (1994)
34. Farah, C. S., Miyamoto, C. A., Ramos, C. H., da Silva, A. C., Quaggio, R. B., Fujimori, K., Smillie, L. B., Reinach, F. C.: Structural and regulatory functions of the NH2- and COOH-terminal regions of skeletal muscle troponin I. J Biol Chem. 269:5230-40. (1994)
35. Tsuda, S., Aimoto, S., Hikichi, K.: 1H-NMR study of Ca(2+)-and Mg(2+)-dependent interaction between troponin C and troponin I inhibitory peptide (Asn96-Met116). J Biochem (Tokyo). 112:665-70. (1992)
36. Pearlstone, J. R., Sykes, B. D., Smillie, L. B.: Interactions of structural C and regulatory N domains of troponin C with repeated sequence motifs in troponin I. Biochemistry. 36:7601-6. (1997)
37. Tripet B, Van Eyk JE, Hodges RS.: Mapping of a second actin-tropomyosin and a second troponin C binding site within the C terminus of troponin I, and their importance in the Ca2+-dependent regulation of muscle contraction. J Mol Biol. 271:728-50. (1997)
38. Van Eyk, J. E., Hodges, R. S.:A synthetic peptide of the N-terminus of actin interacts with myosin. Biochemistry. 30:11676-82. (1991)
39. Slupsky, C. M., Shaw, G. S., Campbell, A. P., Sykes, B. D.: A 1H NMR study of a ternary peptide complex that mimics the interaction between troponin C and troponin I. Protein Sci. 1:1595-603. (1992)
40. Luo, Y., Wu, J-L., Li, B., Langsetmo, K., Gergely J., Tao, T.: Photocrosslinking of Benzophenone-labeled Single Cysteine Troponin I Mutants to Other Thin Filament Proteins. Journal of Molecular Biology, 296:899-910 (2000)
41. Kobayashi, T., Zhao, X., Wade, R., Collins, J. H.: Involvement of conserved, acidic residues in the N-terminal domain of troponin C in calcium-dependent regulation. Biochemistry. 38:5386-91. (1999)
42. O'Neil, K. T., DeGrado, W. F.: How calmodulin binds its targets: sequence independent recognition of amphiphilic alpha-helices. Trends Biochem Sci 15, 59-64 (1990)
43. Vassylyev, D. G., Takeda, S., Wakatsuki, S., Maeda, K. & Maeda, Y.: Crystal structure of troponin C in complex with troponin I fragment at 2.3-Å resolution. Proc. Natl. Acad. Sci. USA. 95, 4847-4852 (1998).
44. Hernandez, G. Blumenthal, D. K. Kennedy, M. A. Unkefer, C. J. Trewhella, J. (1999) Troponin I Inhibitory Peptide (96-115) Has an Extended Conformation When Bound to Skeletal Muscle Troponin C, Biochemistry 38:6911-6917
45. Campbell, A. P., Sykes, B.: Interaction of troponin I and troponin C. Use of the two-dimensional nuclear magnetic resonance transferred nuclear Overhauser effect to determine the structure of the inhibitory troponin I peptide when bound to skeletal troponin C. J Mol Biol 222, 405-21 (1991)
46. McKay, R. T., Pearlstone, J. R., Corson, D. C., Gagne, S. M., Smillie, L. B., Sykes, B. D.: Structure and Interaction Site of the Regulatory Domain of Troponin-C When Complexed with the 96-148 Region of Troponin-I. Biochemistry37, 12419-30 (1998)
47. Li, M. X., Spyracopoulos, L., Sykes, B. D. : Binding of cardiac troponin-I147-163 induces a structural opening in human cardiac troponin-C.Biochemistry. 38, 8289-98 (1999)
48. Meador, W. E., Means, A. R., Quiocho, F. A.: Target enzyme recognition by calmodulin: 2.4 A structure of a calmodulin-peptide complex. Science. 257:1251-5. (1992)
49. Chattopadhyaya, R., Meador, W.E., Means, A.R. & Quiocho, F.A. (1992). Calmodulin structure refined at 1.7 Å. J.Mol. Biol. 228, 1177-1192.
50. Kretsinger, R.H. & Nockolds, C.E. (1973). Carp muscle Ca2+-binding protein. II. Structure determination and general description. J. Biol. Chem. 248, 3313-3326.
51. Ikura, M., et al., & Bax, A. (1992). Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science 256, 632-638.
52. Meador, W.E., Means, A.R. & Quiocho, F.A. (1993). Modulation of calmodulin plasticity in molecular recognition on the basis of X-ray structures. Science 262, 1718-1721.
53. Houdusse, A. & Cohen, C. (1995). Target sequence recognition by the calmodulin superfamily: implications from light chain binding to the regulatory domain of scallop myosin. Proc. Natl. Acad. Sci. USA 92, 10644-10647.
54. Houdusse, A., Silver, M. & Cohen, C. (1996). A model of Ca2+-free calmodulin binding to unconventional myosins reveals how calmodulin acts as a regulatory switch. Structure 4, 1475-1490.
55. Cohen, C. (1975). The protein switch of muscle contraction. Sci. Am. 233, 36-45.
56. Zot, H.G. & Potter, J.D. (1982). A structural role for the Ca2+-Mg2+ sites on troponin C in the regulation of muscle contraction. Preparation and properties of troponin C depleted myofibrils. J. Biol. Chem. 257, 7678-7683.
57. Kuboniwa, H., et al., & Bax, A. (1995). Solution structure of calcium- free calmodulin. Nat. Struct. Biol. 2, 768-776.
58. Zhang, M., Tanaka, T. & Ikura, M. (1995). Ca2+-induced conformational transition revealed by the solution structure of apo calmodulin. Nat. Struct. Biol. 2, 758-767.
59. Gagne, S.M., et al., & Sykes, B.D. (1995). Structures of troponin C regulatory domains in the apo and Ca2+ saturated states. Nat. Struct. Biol. 2, 784-789.
60. Slupsky, C.M. & Sykes B.D. (1995). NMR solution structure of Ca2+- saturated skeletal muscle troponin C. Biochem. 34, 15953-15964.
61. Grabarek, Z., Drabikowski, W., Vinokurov, L. & Lu, R.C. (1981). Digestion of troponin C with trypsin in the presence and absence of Ca2+. Identification of cleavage points. Biochim. Biophys. Acta. 671, 227-233.
62. Potter, J.D. & Gergely, J. (1975). The Ca2+ and Mg2+ binding sites on troponin and their role in the regulation of myofibrillar adenosine triphosphatase. J. Biol. Chem. 250, 4628-4633.
63. Leavis, P.C., Rosenfeld, S.S., Gergely, J., Grabarek, Z. & Drabikowski, W. (1978). Proteolytic fragments of troponin C. Localization of high and low affinity Ca2+ binding sites and interactions with troponin I and troponin T. J. Biol. Chem. 253, 5452-5459.
64. Swindells, M.B. & Ikura, M. (1996). Pre-formation of the semi-open conformation by the apo-calmodulin C-terminal domain and implications for binding IQ-motifs. Nat. Struct. Biol. 3, 501-504.
65. da Silva, A.C., et al., & Reinach, F.C. (1993). Troponin-C mutants with increased Ca2+ affinity. Eur. J. Biochem. 213, 599-604.
66. Fujimori, K., et al., & Reinach, F.C. (1990). Probing the Ca2+-induced conformational transition of troponin C with site-directed mutants. Nature 345, 82-184.
67. Xie, X., et al., & Cohen, C. (1994). Structure of the regulatory domain of scallop myosin at 2.8 A resolution. Nature 368, (6469), 306-312.
68. Houdusse, A. & Cohen, C. (1996). Structure of the regulatory domain of scallop myosin at 2.0 Å resolution: implications for regulation. Structure 4, 21-32.
69. Gulati J., Babu, A., Su, H. & Zhang, Y.F. (1993). Identification of the regions conferring calmodulin-like properties to troponin C. J. Biol. Chem. 268, 11685-11690.
70. Grabarek, Z., Tao, T. & Gergely, J. (1992). Molecular mechanism of troponin-C function. J. Muscle Res. Cell Motil. 13, 383-393.
71. Farah, C.S. & Reinach, F.C. (1995). The troponin complex and regulation of muscle contraction. FASEB J. 9, 755-767.
72. Ingraham, R.H. & Swenson, C.A. (1984). Binary interactions of troponin subunits. J. Biol. Chem. 259, 9544-9548.
73. Wang, C.K. & Cheung, H.C. (1985). Energetics of the binding of Ca2+ and troponin I to troponin C from rabbit skeletal muscle. Biophys. J. 48, 727-739.
74. Grabarek, Z., Drabikowski, W., Leavis, P.C., Rosenfeld, S.S. & Gergely, J. (1981). Proteolytic fragments of troponin C. Interactions with the other troponin subunits and biological activity. J. Biol. Chem. 256, 13121-13127.
75. Farah, C.S., et al., & Reinach, F.C. (1994). Structural and regulatory functions of the NH2- and COOH-terminal regions of skeletal muscle troponin I. J. Biol. Chem. 269, 5230-5240.
76. Matthews, B.W. (1968). Solvent content of protein crystals. J. Mol. Biol. 33, 491-497.
77. Otwinowski, Z. (1993). DENZO: an oscillation data processing program for macromolecular crystallography. Yale University, New Haven, CT.
78. Otwinowski, Z. (1993). SCALEPACK: Software for the scaling together of integrated intensities measured on a number of separate diffraction images. Yale University, New Haven, CT.
79. Cowtan, K.D. (1994). "DM": an automated procedure for phase improvement by density modification. In Joint CCP4 and ESF- EACBM Newsletter on Protein Crystallography 31, 34-38.
80. Otwinowsky, Z. (1991). Proceedings of the CCP4 Study Weekend. (Wolf, W., Evans, P.R. & Leslie, A.G.W., eds), Daresbury Laboratory, Warrington, UK.
81. Brünger, A.T., Kuriyan, J. & Karplus, M. (1987). Crystallographic R- factor refinement by molecular dynamics. Science 235, 458-460.
82. Lamzin, V.S. & Wilson, K.S. (1993). Automated refinement of protein models. Acta Cryst. D 49, 129-147.
83. Jones, T.A., Zou, J.-Y., Cowan, S.W. & Kjeldgaard, M. (1991). Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. A 47, 110-119.
84. Laskowski, R.A., MacArthur, M.W., Moss, D.S. & Thornton, J.M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283-291.
85. Navaza, J. (1994). AMoRe: an Automated Package for Molecular Replacement. Acta Cryst. A 50, 157-163.
86. Bernstein, F.C., et al., & Tasumi, M. (1977). The protein data bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 112, 535-542.
87. Kraulis, P.J. (1991). MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24, 946-950.
88. Brünger, A.T. (1992). The free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 335, 472-474.
89. Kleerekoper Q, Howarth JW, Guo X, Solaro RJ, Rosevear PR. (1995) Cardiac troponin I induced conformational changes in cardiac troponin C as monitored by NMR using site-directed spin and isotope labeling. Biochemistry 34:13343-52
90. Kobayashi T, Zhao X, Wade R, Collins JH. (1999) Ca2+-dependent interaction of the inhibitory region of troponin I with acidic residues in the N-terminal domain of troponin C. Biochim Biophys Acta 1430:214-21
91. Olah GA, Rokop SE, Wang CL, Blechner SL, Trewhella J (1994) Troponin I encompasses an extended troponin C in the Ca(2+)-bound complex: a small-angle X-ray and neutron scattering study. Biochemistry 1994 Jul 12;33(27):8233-9
92. Finley N, Abbott MB, Abusamhadneh E, Gaponenko V, Dong W, Gasmi-Seabrook G, Howarth JW, Rance M, Solaro RJ, Cheung HC, Rosevear PR (1999) NMR analysis of cardiac troponin C-troponin I complexes: effects of phosphorylation. FEBS Lett 453:107-12
93. Brunger A.T., Adams P.D., Clore G.M., Delano W.L., Gros P., Grosse-Kuntsleve R.W., Jiang J.-S., Kuszewski J., Nilges N., Pannu N.S., Read R.J., Rice L.M., Simonson T., Warren G.L. (1998) Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Cryst. D54, 905-921
94. Trewhella J, Blumenthal DK, Rokop SE, Seeger PA. (1990) Small-angle scattering studies show distinct conformations of calmodulin in its complexes with two peptides based on the regulatory domain of the catalytic subunit of phosphorylase kinase. Biochemistry 29:9316-24
95. Sia SK, Li MX, Spyracopoulos L, Gagne SM, Liu W, Putkey JA, Sykes BD. (1997) Structure of cardiac muscle troponin C unexpectedly reveals a closed regulatory domain. J Biol Chem 272:18216-21
96. Sundaralingam M, Bergstrom R, Strasburg G, Rao ST, Roychowdhury P, Greaser M, Wang BC. (1985) Molecular structure of troponin C from chicken skeletal muscle at 3-angstrom resolution. Science 227:945-8
97. Herzberg O, James MN. (1985) Structure of the calcium regulatory muscle protein troponin-C at 2.8 A resolution. Nature 313:653-9
98. Sundaralingam M, Drendel W, Greaser M. (1985) Stabilization of the long central helix of troponin C by intrahelical salt bridges between charged amino acid side chains. Proc Natl Acad Sci 82:7944-7
99. Tao T, Gong BJ, Leavis, PC, (1990) Calcium-induced movement of troponin-I relative to actin in skeletal muscle thin filaments. Science 247:1339-1341
100. Tao T, Gowell E, Stasburg GM, Gergely J, Leavis, PC, (1989) Ca2+ dependence of the distance between Cys-98 of troponin C and Cys-133 of troponin I in the ternary troponin complex: resonance energy transfer measurements. Biochemistry 28:5902-5908
101. Ramakrishnan S, Hitchcock-DeGregori SE. (1996) Structural and functional significance of aspartic acid 89 of the troponin C central helix in Ca2+ signaling. Biochemistry 35:15515-21
102. Luo Y, Wu JL, Gergely J, Tao T. (1998) Biophys J 74:3111-9 Localization of Cys133 of rabbit skeletal troponin-I with respect to troponin-C by resonance energy transfer.
103. Ikura M, Clore GM, Gronenborn AM, Zhu G, Klee CB, Bax A. (1992) Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science 256:632-8
104. Mirzoeva S, Weigand S, Lukas TJ, Shuvalova L, Anderson WF, Watterson DM. (1999) Analysis of the functional coupling between calmodulin's calcium binding and peptide recognition properties. Biochemistry 38:3936-47
105. Kimura A, Harada H, Park JE, Nishi H, Satoh M, Takahashi M, Hiroi S, Sasaoka T, Ohbuchi N, Nakamura T, Koyanagi T, Hwang TH, Choo JA, Chung KS, Hasegawa A, Nagai R, Okazaki O, Nakamura H, Matsuzaki M, Sakamoto T, Toshima H, Koga Y, Imaizumi T, Sasazuki T. (1997) Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet. 16:379-82.
106. James J. et al. (1999) Circulation 100:I-276
107. Murphy AM, Kögler H, Georgakopoulos D, McDonough JL, Kass DA, Van Eyk JE, Marbán E. (2000) Transgenic Mouse Model of Stunned Myocardium. Science 287:488-491 Dobrowolski, Z., Xu, G. Q., Hitchcock-DeGregori, S. E.: Modified calcium-dependent regulatory function of troponin C central helix mutants. J Biol Chem. 266:5703-10. (1991)
109. Babu, A., Rao, V. G., Su, H., Gulati, J.: Critical minimum length of the central helix in troponin C for the Ca2+ switch in muscular contraction. J Biol Chem. 268:19232-8. (1993)
110. Ramakrishnan, S., Hitchcock-DeGregori, S. E.: Investigation of the structural requirements of the troponin C central helix for function. Biochemistry. ;34:16789-96. (1995)
111. Ramakrishnan, S., Hitchcock-DeGregori, S. E.: Structural and functional significance of aspartic acid 89 of the troponin C central helix in Ca2+ signaling. Biochemistry. 35:15515-21. (1996)
112. Soman, J., Tao, T., Phillips, G. N. Jr.: Conformational variation of calcium-bound troponin C. Proteins. 37:510-1. (1999)
113. Takahashi-Yanaga F, Morimoto S, Ohtsuki I.: Effect of Arg145Gly Mutation in Human Cardiac Troponin I on the ATPase Activity of Cardiac Myofibrils. J Biochem (Tokyo). 127:355-357. (2000)
114. Li Y, Love ML, Putkey JA, Cohen, C.: Bepridil opens the regulatory N-terminal lobe of cardiac troponin C. PNAS 97:5140-5145. (2000)
115. Merritt, E.A. & Bacon, D.J.: Raster3D Photorealistic Molecular Graphics. Methods in Enzymology 277, 505-524. (1997)
116. MOLSCRIPT (see reference 87) as modified by Peisach D & Peisach E. http://cerebus.biochem.med.umich.edu/~peisach/povscript/.
117. Sayle R, Milner-White EJ.: RasMol: Biomolecular graphics for all. TIBS. 20:374. (1995)
118. Wang ZY, Sarkar S, Gergely J, Tao T.: Ca2(+)-dependent interactions between the C-helix of troponin-C and troponin-I. Photocross-linking and fluorescence studies using a recombinant troponin-C. J Biol Chem. 265:4953-7. (1990)